Molecular mechanisms of amyotrophic lateral sclerosis (ALS) and related neurodegenerative disorders
Amyotrophic lateral sclerosis (ALS) is an adult-onset progressive neuromuscular disease affecting motor neurons in the spinal cord and motor cortex. Up to 90% of ALS cases are sporadic (sALS), with the remaining 10% caused by a mutation(s) in known genes (familial ALS, fALS). ALS is inevitably fatal (usually within 3-5 years of diagnosis), currently with no cure. Molecular mechanisms shared by different fALS subtypes and by fALS and sALS are still poorly understood which represents the major obstacle in establishing therapeutic targets for this devastating disease.
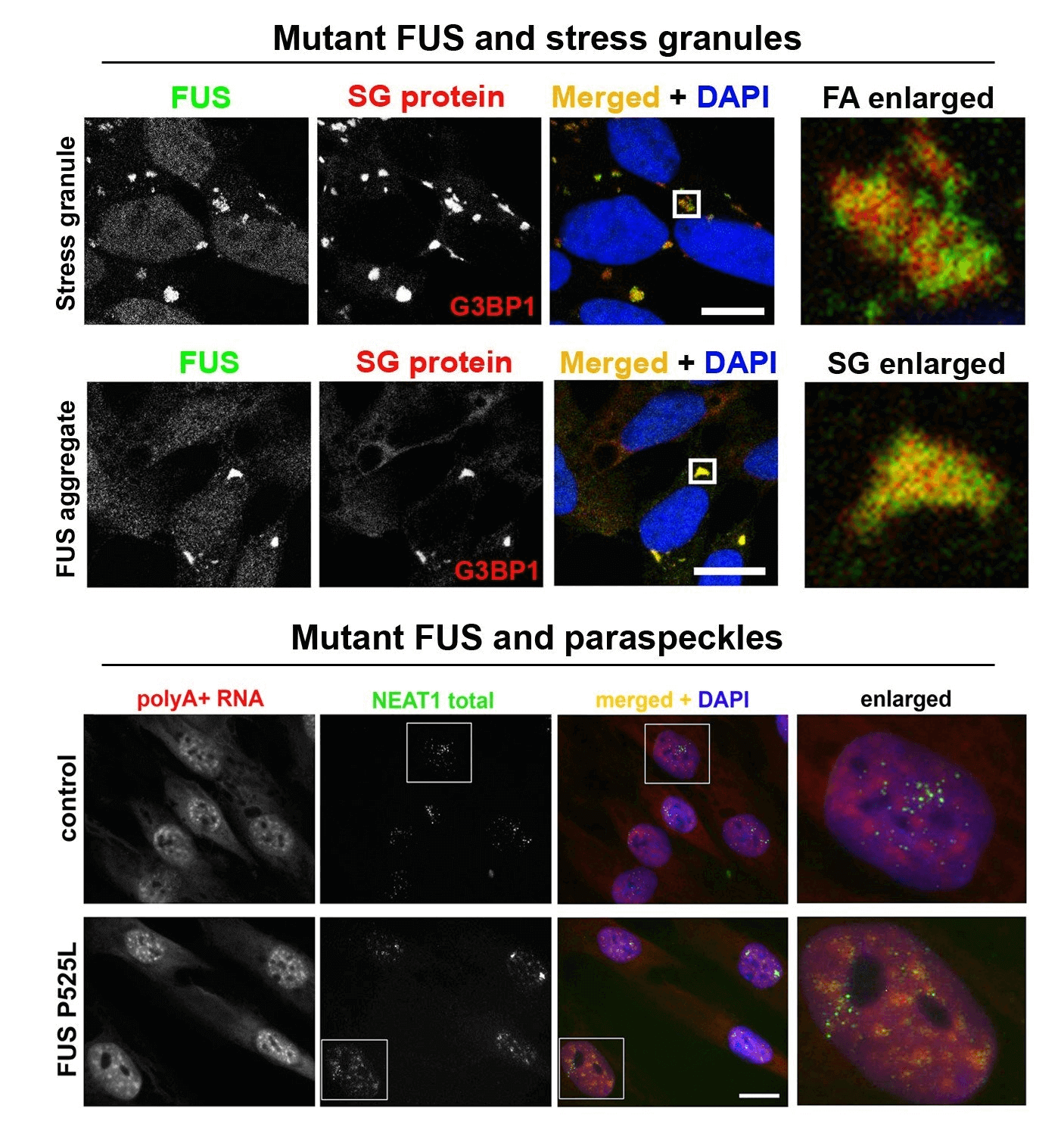
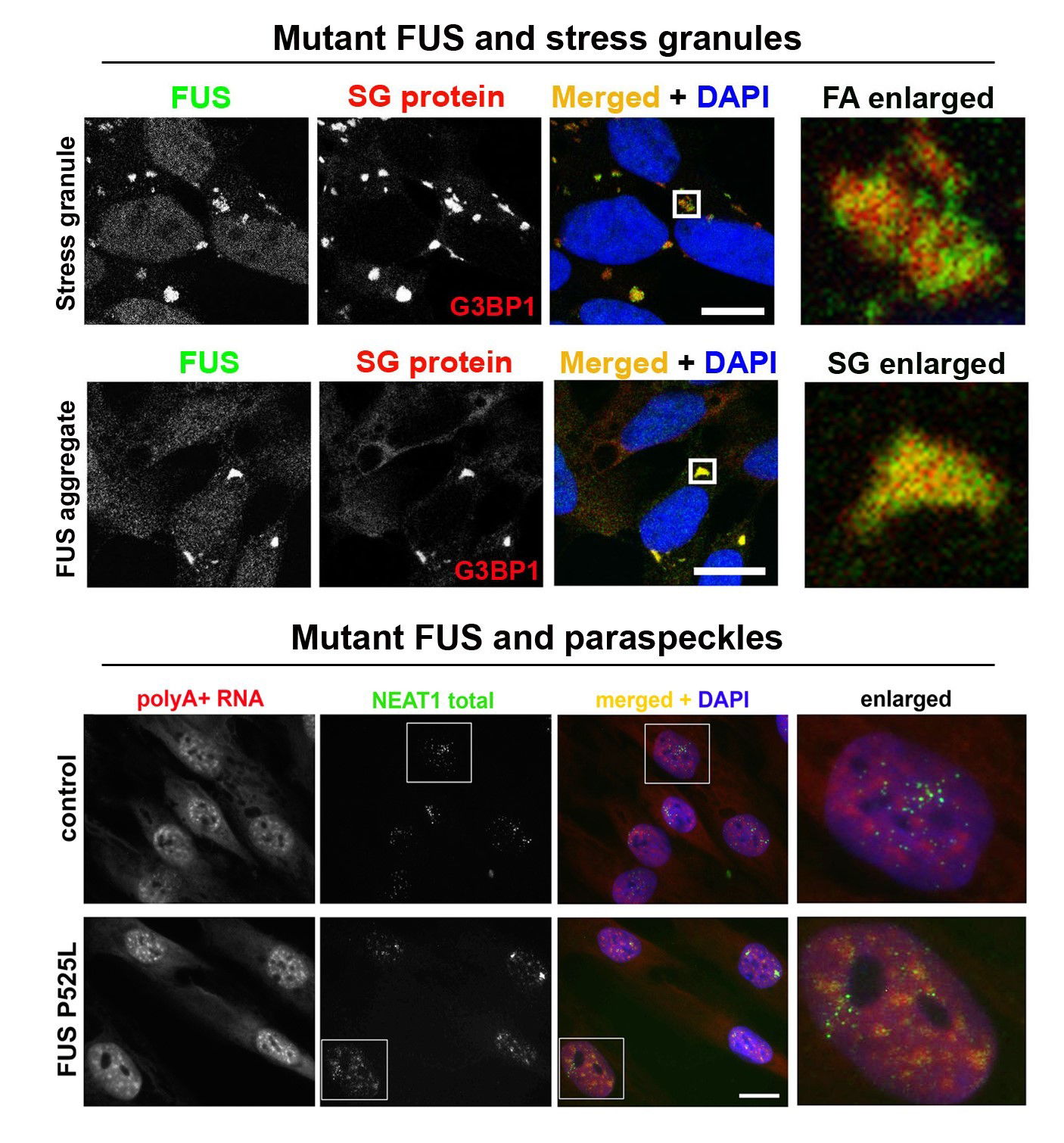
LncRNA NEAT1 and paraspeckle dysfunction in ALS. Nuclear enriched abundant transcript 1 (NEAT1) is a long non-coding RNA (lncRNA) which is known to regulate gene expression and multiple aspects of cellular stress response. In particular, NEAT1 contributes to regulation of transcription and chromatin active state. NEAT1 has long been known to be dysregulated in many cancers and in some autoimmune conditions, however more recently, this lncRNA has also been implicated in normal neuronal functions as well as in the pathophysiology of neurological conditions. Expression of NEAT1 in the nervous system is altered in many neurodegenerative diseases including frontotemporal dementia, Alzheimer’s disease, Parkinson’s disease and ALS (for review, see An, Williams, Shelkovnikova, 2018). Among neurodegenerative diseases, the strongest experimental and histopathological evidence links NEAT1 and ALS pathogenesis. Analysis of post-mortem tissue from ALS patients demonstrated that accumulation of NEAT1_2 is prominent in survived neurons in the spinal cord indicating that its upregulation in this neuronal subtype is likely protective (Shelkovnikova et al., 2018; An et al., 2019). NEAT1_2 accumulation might be the much-sought ‘universal’ molecular signature shared by aetiologically different ALS cases and therefore represent an attractive drug target. Increasing NEAT1_2 levels in motor neurons in ALS can be used to preserve their function for longer, delaying the disease onset in inherited ALS cases and preventing the decline in muscle strength and improving survival in sporadic ALS cases.
We use three different model systems to study NEAT1 roles in ALS pathogenesis:
- Cell models (stable cell lines, human stem cell derived motor neurons and patients’ cells)
We have various stable cell lines with knockout of one or both NEAT1 isoforms, and NEAT1 KO hES cells are currently in production.
- Transgenic Drosophila
We actively collaborate with a group at the University of Tokyo (Dr Hashimoto) who are experts in ALS modeling in Drosophila.
- Transgenic mice
We have access to NEAT1 knockout mouse line through collaboration with our Japanese colleagues (our recent preprint on the neurological phenotypes in these mice can be found here). We also generated and are currently characterising NEAT1 transgenic (overexpressing) mice in collaboration with IPAC RAS (Russian Federation).
Abnormal metabolism of RNA-binding proteins in ALS. A significant proportion of ALS cases is caused by mutations in RNA-binding proteins (RBPs) and/or involves their functional dysregulation which points to abnormal RNA metabolism is a common pathogenetic mechanism. Most RBPs dysregulated in ALS are components of a class of membraneless multiprotein complexes called RNA (RNP) granules that includes stress granules, paraspeckles, neuronal RNA granules, Gems and P-bodies, among others. These RBPs possess so called low-complexity (LC), intrinsically disordered domains in their structure which are responsible for their ability to phase-separate and to be recruited into RNP granules in the nucleus and cytoplasm. The same domains drive pathological aggregation of these proteins. A growing list of RNP granule proteins have been implicated in ALS, most of them are modified by disease-causative mutations, including TDP-43, FUS, TAF15, EWS, hnRNP A2/B1, hnRNP A1, TIA-1, profilin1, angiogenin, SFPQ and CREST. We are focusing on FUS and TDP-43 proteins, which are both components of stress granules and paraspeckles and play important roles in the regulation of these two RNP granules. In particular, we recently found that TDP-43 and FUS loss and gain of function can contribute to ALS development by affecting NEAT1/paraspeckles (Shelkovnikova et al., 2018; An et al., 2019). We are also interested in external (environmental) factors, or “secondary hits”, in ALS pathogenesis. Highlights from our recent study linking antiviral immune response and ALS-FUS are shown in Figure 1 and can be found here. We are also interested in an ALS-linked protein CREST (latest collaborative research paper can be found here). As tools in our everyday research, we use CRISPR/Cas9 cell lines, patient-derived fibroblasts as well as a vast collection of plasmids for transient expression.
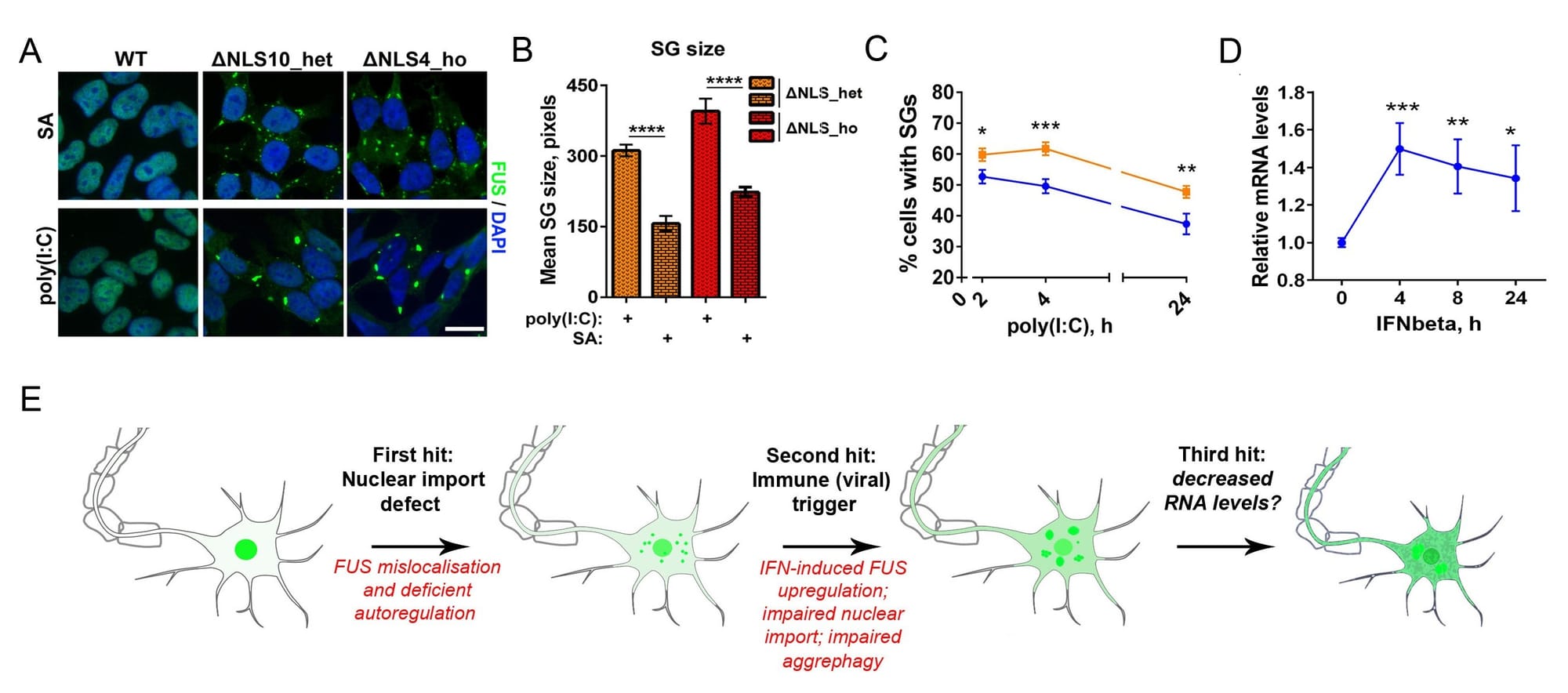
Figure 1. Mimicking viral infection in mutant FUS expressing cells can promote FUS pathology (proteinopathy). (A-C) Viral dsRNA mimic causes formation of large, persistent stress granules in cells expressing cytoplasmic mutant FUS (FUS CRISPR/Cas9 cell lines). (D) Major component of antiviral immune response, IFNbeta, increases FUS expression. (E) Model of FUS proteinopathy development in ALS-FUS.


