Therapeutic targeting of RNA
Many RNA molecules represent attractive drug targets. However, RNAs are traditionally thought to be difficult to target using small molecules. It is now increasingly appreciated that despite linearity and limited structural complexity, most RNAs contain complex 3D motifs in their structure that can serve as platforms for small molecule binding. We are exploring the possibility of using small molecules for targeting of protein-coding and non-coding RNAs to expand the boundaries of the druggable genome.
RNA – not undruggable anymore? Multiple human diseases are characterised by altered RNA metabolism. A prominent example is neurodegenerative diseases caused by microsatellite repeat expansions, e.g. myotonic dystrophy, Huntington's disease, spinocerebellar ataxias and some forms of motor neuron disease, all characterised by accumulation of toxic RNA species. More than 70% human genome gives rise to non-coding RNAs, and the number of disease-linked non-coding RNAs is constantly growing. Several classes of regulatory non-coding RNAs are implicated in common neurodegenerative diseases, including tRNAs, miRNA and lncRNAs, e.g. BACE1 antisense transcript in Alzheimer’s disease, pre-miR17~92 in cancer and motor neuron disease or long non-coding RNA MALAT1 in cancer. Many of these RNA molecules would represent attractive drug targets. Furthermore, only 10-15% disease-linked proteins belong to the druggable protein families, such as kinases and GPCRs, whereas the remaining 85% do not possess convenient small molecule pockets or do not fold into defined/stable conformation. The ability to target these proteins at the level of their pre-mRNAs or mRNAs would also significantly expand the druggable genome. RNA splicing correction approaches deserve a particular attention in this regard because they provide an exciting opportunity to correct both primary genetic defects and downstream pathogenic events in a variety of diseases. For example, the success of two splicing modulators branaplam and risdiplam, which are currently in Phase 2 clinical trials for a severe childhood disorder spinal muscular atrophy (SMA), resurrected the hopes of translational researchers for finding efficient small molecules working at the level of RNA. Overall, the ability to target RNAs would completely change the concept of druggability.
Targeting RNA with small molecules. Medicinal chemists at the MDI are experts in the development of therapeutic small molecules. Small molecules with their good pharmacokinetic profile provide an alternative to the use of antisense oligonucleotides (ASOs) traditionally viewed as the best means to target RNA therapeutically. Despite linearity of the RNA molecule and the limited structural complexity of its primary structure, most protein-coding and non-coding RNAs contain complex 3D motifs in their structure characterised by predictable shape and most importantly, behaviour. Such 3D structural motifs, e.g. stem-loops, tetra-loops, helices, bulges and their combinations, can become a platform for small molecule binding, akin to small molecule pockets in proteins. Small molecule binding to RNAs can modulate their biology by affecting complexes of this RNA with the binding protein partners or by changing RNA stability. Importantly, the RNA target does not have to be globally folded, rather, short structured sequences can provide sufficient binding sites for small molecules and can be harnessed to efficiently modulate stability, interactions and function of this RNA.
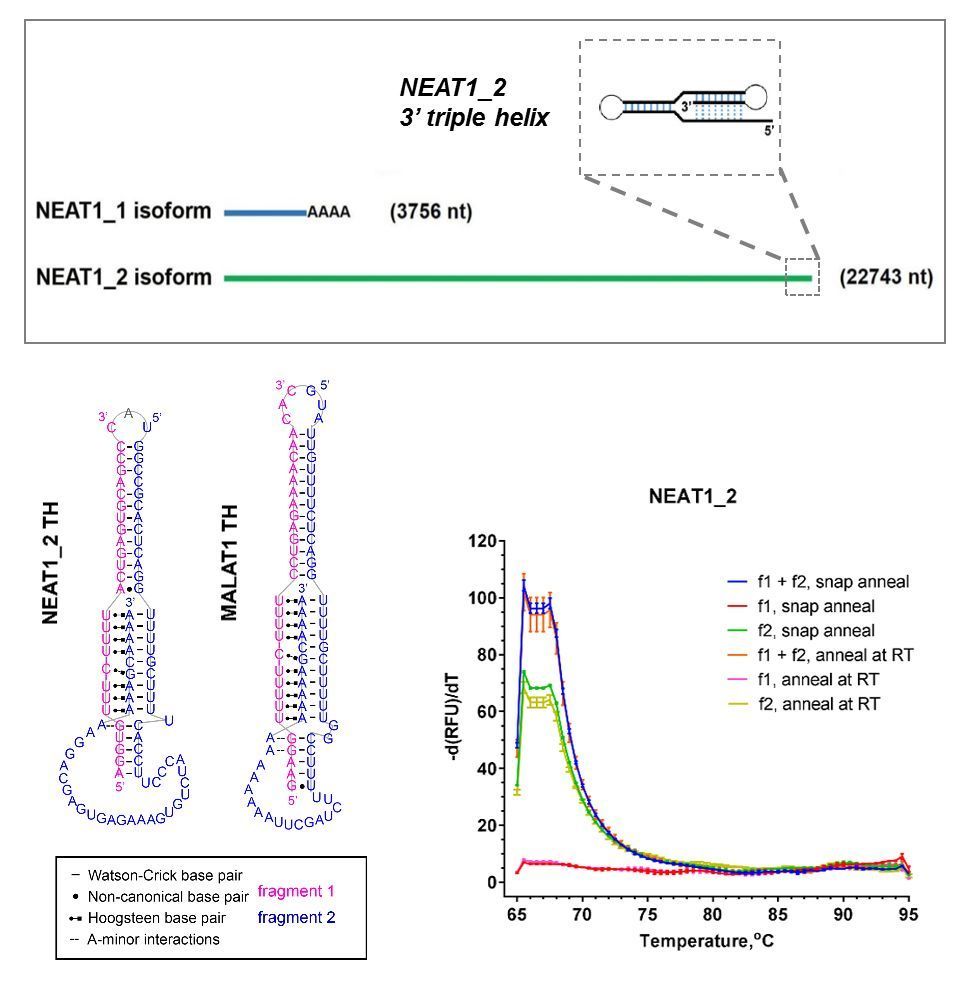
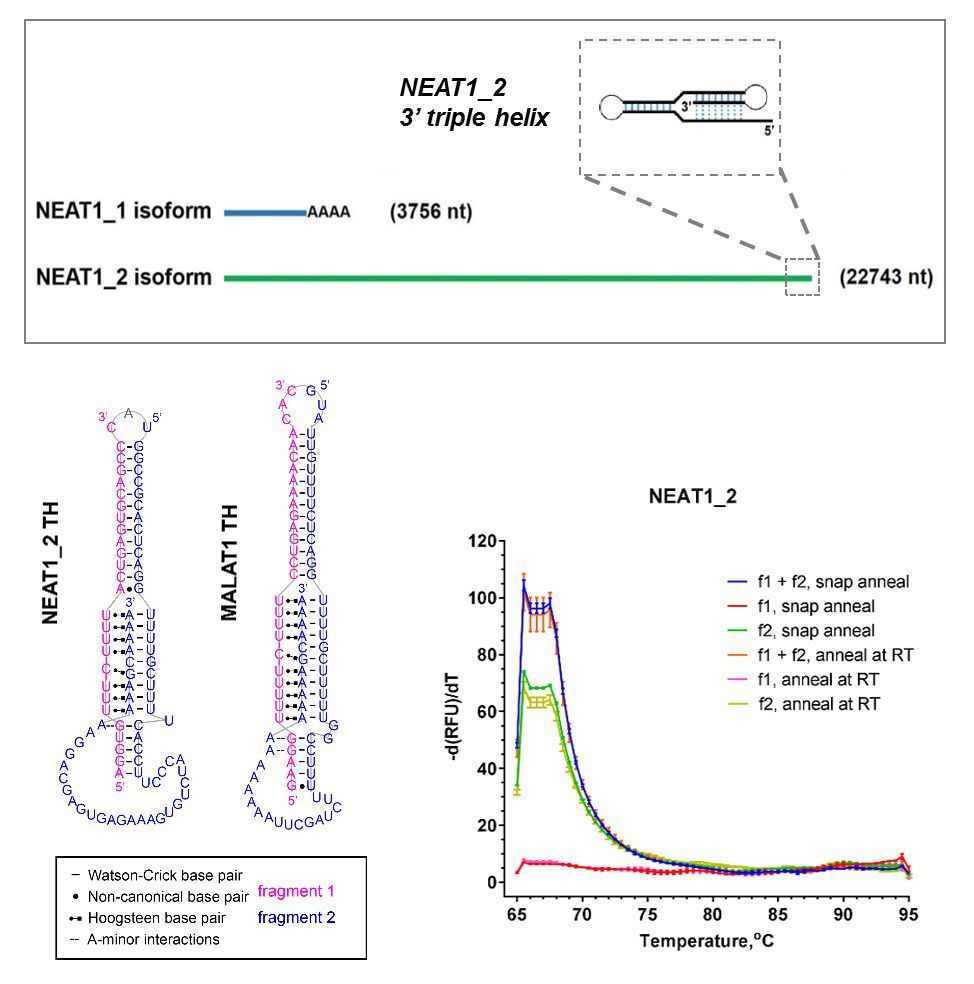
NEAT1_2. Nuclear Enriched Abundant Transcript 1 (NEAT1) is a ubiquitous, highly expressed nuclear-retained regulatory long non-coding RNA (lncRNA) playing important roles in human physiology and pathophysiology. Considered one of the most therapeutically important non-coding RNAs, NEAT1 has long been known to play an important role in cancer. NEAT1 gene is a hotspot for mutations in some types of cancer, and NEAT1 is overexpressed in the majority of solid tumours, for example breast and prostate cancer. High NEAT1 levels in tumour cells predict poor outcome, recurrence and therapy resistance. Silencing NEAT1 arrests solid tumour growth in mice. Yet mounting evidence also suggests that abnormal NEAT1 expression contributes to neurological diseases. In particular, NEAT1 is among the most dysregulated transcripts in the brain tissue in neurodegenerative diseases, including frontotemporal dementia, motor neuron disease, Alzheimer's, Huntington's and Parkinson’s diseases.
The NEAT1 locus produces two transcripts, NEAT1_1 and NEAT1_2, and the longer NEAT1 isoform, NEAT1_2, is essential for the assembly of nuclear RNP granules paraspeckles. Multiple reports suggest that NEAT1_2 is the primary NEAT1 isoform driving cancer progression. Among neurodegenerative diseases, the strongest experimental and histopathological evidence links NEAT1_2 and ALS pathogenesis. Analysis of post-mortem tissue from ALS patients demonstrated that accumulation of NEAT1_2 is prominent in survived neurons in the spinal cord indicating that its upregulation in this neuronal subtype is likely protective (Shelkovnikova et al., 2018; An et al., 2019). NEAT1_2 accumulation might be the much-sought ‘universal’ molecular signature shared by aetiologically different ALS cases and therefore an attractive drug target. Increasing NEAT1_2 levels in motor neurons in ALS can be used to preserve their function for longer, delaying the disease onset in inherited ALS cases and preventing the decline in muscle strength and improving survival in sporadic ALS cases.
Currently no selective NEAT1-modyfing drugs are available. NEAT1_2 contains an element for nuclear expression (ENE) – a short (93 nt) RNA triple helix motif of sufficient complexity which is potentially amenable to small molecule targeting. Intact NEAT1_2 triple helix is critically important for the molecule’s stability, and therefore chemicals selectively binding to this motif can be used to modulate NEAT1_2 levels. Identification of chemotypes capable of binding NEAT1_2 and promoting its degradation has a potential to give rise to a novel class of anticancer therapeutics, whereas drugs which can selectively stabilise it will hold promise for ALS and other neurodegenerative diseases. In the lab, we are developing HTS-compatible in vitro (Figure 1) and cellular (Figure 2) assays to enable the discovery of new chemical ligands for this ncRNA.
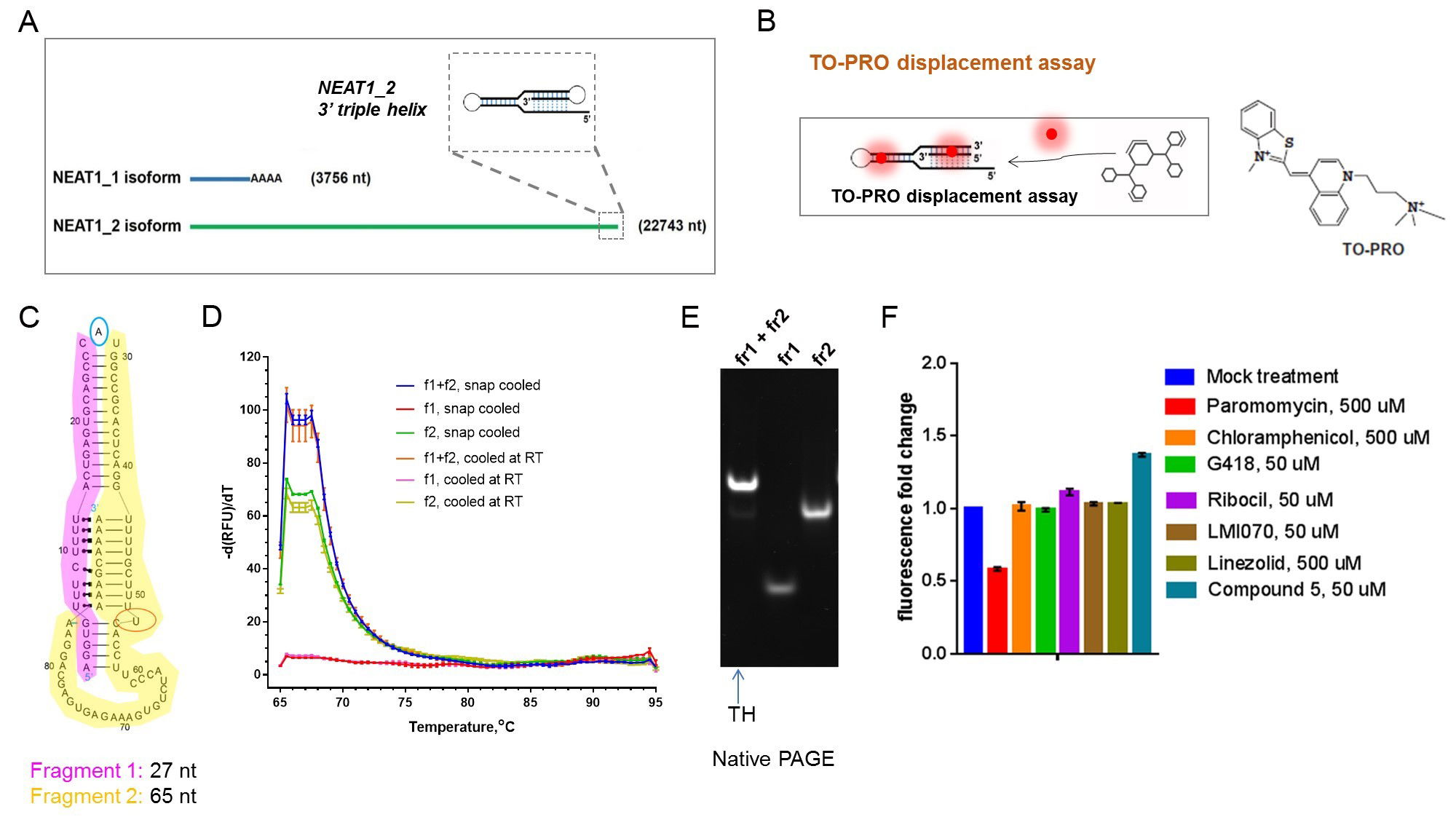
Figure 1. In vitro assay development for NEAT1_2 triple helix (TH). (A) NEAT1 isoforms and the position of the ENE (triple helix) in NEAT1_2. (B) Principle of TO-PRO based fluorescent displacement assay. (C) Bipartite NEAT1_2 TH used in the assay development. (D,E) Characterisation of the bipartite TH by melting analysis (D) and native PAGE (E). (F) Effect of RNA-binding compounds on the fluorescence of TO-PRO bound to NEAT1_2 TH.
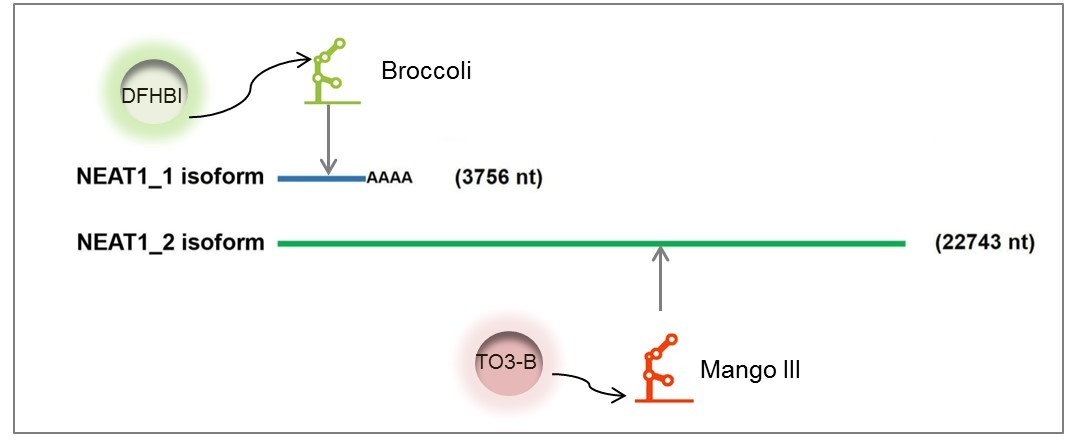
Figure 2. Generation of reporter cell lines with aptamer-labelled endogenous NEAT1 for cellular assay development.
Splicing targets. We are also interested in splicing targets, in particular for ALS and related disorders. The RNA-binding protein TDP-43 is known to be depleted and aggregated in ~90% of ALS cases. TDP-43 is a splicing factor which regulates a multitude of targets in neurons. In particular, TDP-43 loss of function results in an abnormal splicing pattern in a pre-mRNA encoding a microtubule-associated protein essential for axonal integrity and regeneration. Within this project, an HTS-compatible reporter cell line is being developed which will inform of the restoration of splicing pattern within this pre-mRNA by small molecules in the absence of TDP-43. A cellular assay established on the basis of this reporter cell line will be used to identify chemical starting points to develop small molecule splicing modulators for this therapeutic target.


